药智新闻2023-11-29 17:32

![]()
非酒精性脂肪性肝炎(NASH),是近年来行业公认的待解决疾病,拥有广泛的患病人群和医药市场需求。
据弗若斯特沙利文预测,到2025年全球NASH药物市场将>100亿美元,并于2030年>300亿美元。当前,已有数百个药物进入到临床开发,但尚未有品种获FDA批准。
在挺进NDA和临床III期的品种当中,笔者对热门品种进行重点信息梳理,以最快、最直观的展现NASH领域的重点品种特点。
01
“意难平”的奥贝胆酸
奥贝胆酸是InterceptPharmaceuticalsInc开发的一款FXR激动剂,最早于2016年获美国FDA批准上市,适应症为胆汁性肝硬化,后于EMEA批准用于原发性胆汁性胆管炎的治疗。
对于NASH适应症的开发,奥贝胆酸在该领域处于第一梯队。早在2015年,该品种就在适应症NASH肝纤维化方面获得FDA的突破性疗法认定,且是第一个在NASH领域进入到临床III期的品种,并进展到美国FDA方面的注册申请。但遗憾的是,在今年上半年,开发公司Intercept官宣FDA再次拒绝该品种的注册申请,并在回复函中提到“在NASH中重新提交的NDA至少需要成功完成REGENERATE研究的长期临床结果”,多次负面结果的叠加也导致了奥贝胆酸在NASH领域的阶段性研究暂停。
进一步从数据来看,早期REGENERATE公开的数据显示:治疗18个月时,奥贝胆酸高剂量组中23%的患者实现了纤维化改善≥1级且NASH无恶化,而安慰剂组仅达到12%,但高剂量组瘙痒发生率高达51%,而安慰剂组仅为19%;同时奥贝胆酸会导致LDL-C升高,以及存在潜在的心血管风险。二次中期数据分析显示,高剂量组有22.4%患者的纤维化改善≥1级且NASH无恶化,安慰剂组为9.6%,但高剂量组瘙痒发生率55%,安慰剂组为24%。
图1FDA拒绝&Intercept暂停NASH投资
图片来源:https://firstwordpharma.com/story/5754177
02
瑞司美替罗挺进NDA
行业关注度较高的THRβ激动剂瑞司美替罗(Resmetirom),已于2023年7月开展了注册申请工作,是目前最具潜力获FDA批准的NASH品种。
瑞司美替罗的开发公司为Madrigal,最初由罗氏开发,并于2016年开展相关的NASH研究,2019年于多个国家开展III期临床,并主推NASH。2023年上半年,该品种获FDA突破性疗法认定,2023年7月进行注册申请,适应症为“非酒精性脂肪性肝炎伴肝纤维化”,且当前已纳入优先审评。
为了推进该品种在NASH适应症的突破,Madrigal已经开展4个III期临床,以证明该品种的安全有效。2023年11月,Madrigal公开了MAESTRO-NASH的部分试验结果:给药100mg瑞司美替罗,约70%患者存在≥30%的MRI-PDFF,而≥30%MRI-PDFF与NASH的缓解(96%的患者)和纤维化的改善(88%的患者)关联度极高。该组MRI-PDFF总体中位降低率为52%。另,该品种耐受性方面,主要为轻度和短暂性腹泻以及轻度恶心。
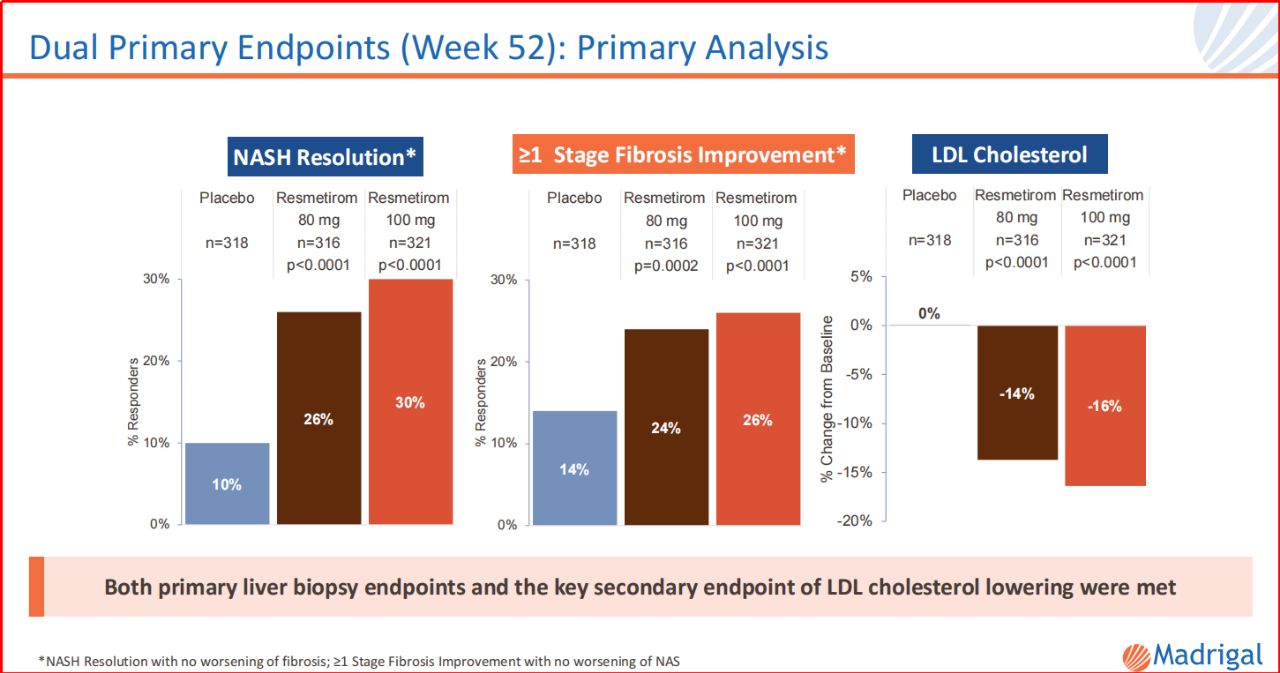
图2MAESTRO-NASH主要终点分析
图片来源:https://ir.madrigalpharma.com/static-files/e8a35f47-f841-49d4-9c21-781f41177609
03
“火出圈”的司美格鲁肽
Semaglutide,凭借在GLP-1领域的大放异彩,已经成为整个药物研发领域的全明星品种,热度不亚于曾经的O药、K药,临床主要用于治疗2型糖尿病和肥胖。
2020年12月,司美格鲁肽在NASH领域的药物研发已经进入临床III期,且相继于多个国家和地区启动临床(包括中国)。2023年3月,还启动了相关非酒精性脂肪性肝炎相关肝硬化的临床工作。
II期临床NN9931-4296(NCT02970942)研究结果:Semaglutide(0.1mg、0.2mg、0.4mg)VS安慰剂组,72周NASH病理消退且肝纤维化未进展的患者比例为40%、36%、59%vs17%,达主要终点。PS:受试人群为NASH合并肝纤维化(F1-3期)。
另,司美格鲁肽还启动了与FXR激动剂cilofexor/ACC抑制剂firsocostat联用的工作,结果显示:司美格鲁肽单药组观察到NASH和肝纤维化的改善,联合组进一步提升疗效;且无论是单药组还是联合用药组均表现了较好的耐受性。
在中国,NASH方面的临床试验为CTR20211818,题目为“Semaglutide对非肝硬化性非酒精性脂肪性肝炎受试者的影响”,主要终点为“脂肪性肝炎缓解且肝纤维化无恶化(是/否)-72周”、“肝纤维化改善且脂肪性肝炎无恶化(是/否)-72周”和“至首例肝脏相关临床事件的时间(复合终点)-240周”。
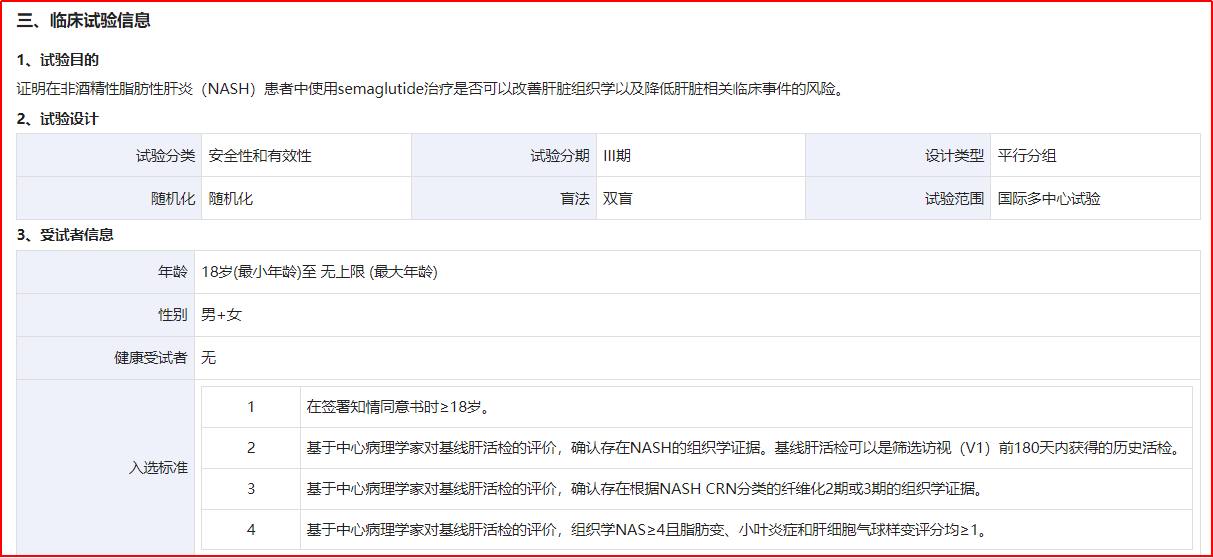
图3Semaglutide-CTR20211818试验信息
图片来源:http://www.chinadrugtrials.org.cn/clinicaltrials.searchlistdetail.dhtml
04
Lanifibranor,正大天晴重仓开发
PPAR激动剂家族,是NASH药物开发的重要组成,当前国内外关注度较高的应为Lanifibranor,以开发NASH适应症为首要方向。该品种的原研公司为Inventiva,后正大天晴药业参与到该品种的开发当中,当前已进入到临床III期,且分别于美国、中国获得突破性疗法认定。
该品种II期临床数据显示:受试者为伴有肝脂肪变性和中重度坏死性炎症且无肝硬化的NASH患者,给药lanifibranor1200mg,试验组脂肪变性活动性纤维化(SAF)活动度评分至少降低2分且肝纤维化不恶化的患者比例显著高于安慰剂组。另,受试者为伴2型糖尿病患者,给药lanifibranor800mg,试验组肝脏脂肪含量相比安慰剂组显著下降。试验期间,lanifibranor的安全性显示胃肠道事件的发生率增加(如腹泻和恶心),以及外周水肿。
国内,Lanifibranor共登记两项临床试验,分别为“评价Lanifibranor单次和多次给药的药代动力学特征和安全性(CTR20233135)”和“评价lanifibranor治疗无肝硬化、伴2期(F2)/3期(F3)肝纤维化的非酒精性脂肪性肝炎(NASH)有效性和安全性(CTR20232876)”。
其中,CTR20232876为一项III期临床,目的为“旨在评价lanifibranor在NASH伴肝纤维化成人受试者中的作用。主要队列两个周期的主要目的是:双盲安慰剂对照(DBPC)期(A部分)评估与安慰剂相比,lanifibranor在NASH缓解同时经肝组织学评估纤维化改善方面的效果。双盲试验药扩展(ATE)治疗期(B部分)评估DBPC期后lanifibranor的安全性。”

图4Lanifibranor-CTR20232876临床终点设定
图片来源:http://www.chinadrugtrials.org.cn/clinicaltrials.searchlistdetail.dhtml
05
试验设计&终点选择,
是机会也是挑战!
近年来,NASH药物开发的大环境已经发生了重大变化,特别要关注监管方的动向。2021年1月,FDA提出了这类药物获批的另一种途径,即申办方需要进行2项III期试验,一项是在高危NASH人群中使用肝脏组织学作为替代终点,另一项是在代偿性肝硬化人群中使用主要不良事件作为终点,来共同证明有效性和安全性,基于此将可能获得对伴有F2-F4纤维化NASH的完全批准。因此,一些申办方开始尝试并更改临床计划及终点设定。
而我国,2019年CDE发布了《非酒精性脂肪性肝炎治疗药物临床试验技术指导原则(试行)》,并明确重点指标评价“有效性评价终点包括临床结局终点和肝组织学替代终点,以及血清生化检查、影像学检查等其他探索性终点”。
另,对于确证临床获益的试验,鉴于组织学改善的替代终点与临床结局的相关性尚未确立,应进行临床试验确证临床结局的获益。推荐以下复合终点:出现失代偿事件(腹水、食管胃底静脉曲张出血或肝性脑病)、肝移植、MELD评分≥15、肝细胞癌、全因死亡。对于NASH无肝硬化的患者,还包括进展至肝硬化。
表1终点指标评价方法及具体内容
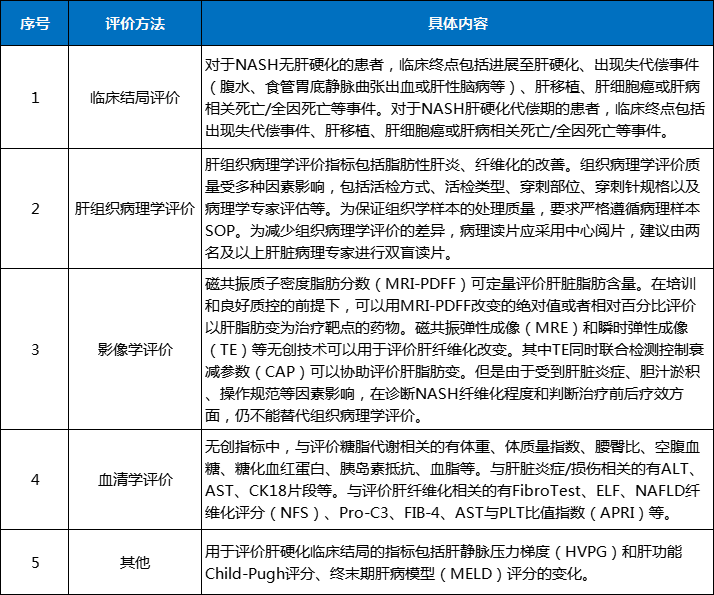
数据来源:CDE
06
小结
总体来看,当前最具潜力的当属THRβ激动剂Resmetirom,但GLP-1司美格鲁肽和正大天晴押宝的Lanifibranor同样值得期待,其他靶点如FGF21、ACC、CCR2/5自然也少不了该领域研发人员的持续关注,尤其是药物联用方面。
另,从NASH适应症多年的药物研发来看,临床试验设计、终点选择、评价标准等,如何科学、客观、有力的证明药物安全有效,需要申办方深度思考,以从源头避免出现意难平。
来源:药智新闻
作者:药智网/中华小吃
 分享
分享

 京公网安备 11010802028547号
京公网安备 11010802028547号
 购物车
购物车





